
Evaluation of Oro-maxillofacial Changes in Major Thalassemia
Ashraf Sayyedi, DDS, MS; Fereydoun Pourdanesh, DDS, MS; Bahador Sarkari, PhD; and Sayyed Hesamedin Nabavizadeh, MD, MS
Commentary by Howard E. Strassler, DMD
Abstract
BACKGROUND: Thalassemia syndromes are the most common single-gene abnormality. Homozygote form (or thalassemia major) is the most severe form of congenital hemolytic anemia in Iran. This disease induces age-related hard and soft tissue changes. AIM: The aim of this study was to evaluate oral and maxillofacial changes in thalassemic major patients who have not had an intensive blood transfusion program, and the prevalence of these changes in different age groups. METHODS AND MATERIALS: This descriptive cross-sectional study was performed on 119 blood-recipient thalassemic patients under the age of 28 by using a questionnaire and performing a clinical examination. Based on research objectives, the patients were divided into six groups. Factors such as mono-maxillary or bi-maxillary protrusions, spacing, gingival and mucous discoloration, tooth color, saddle nose, anterior and posterior malocclusion, and lip incompetency were investigated in all six groups. RESULTS: Results showed that maxillary protrusion, gingival discoloration, saddle nose, and lip incompetency increased with age. Protrusion of the mandible was not common in these patients. The pattern of discoloration of mucosa and anterior malocclusion showed an ascending order until the age of 9 and descended afterward. CONCLUSION: The appearance of oral and maxillofacial signs in thalassemic patients varied according to the patient’s age. Osteal signs appeared early in life if patients did not have an intensive blood-transfusion program or bone-marrow transplantation.
Cooley’s anemia, which is also known as beta (β)-thalassemia or Mediterranean anemia, is a blood disease characterized by malformation of the skull and long bones, which results in an atypical appearance.1 This anemia was first described in 1929 by an American pediatrician.2 It is a disease affecting the synthesis of alpha-(α) or β-globulin chains that make up hemoglobin. Unequal synthesis of globulin chains result in the precipitation of over-synthesized chains, which in turn affects the maturation of red blood cells.3 Gene distribution of thalassemia in the Mediterranean area (Saudi Arabia, Turkey, India, Iran, and Southeast Asia) is 2.5% to 15%. Its prevalence rate in Iran is 10% in the coastal area and 4% to 8% in other areas.4
β-thalassemia is a disease that affects most of the body organs. Orofacial characteristics of this anemia result from the expansion of bone marrow, causing skull and facial deformities. This expansion causes clinically recognizable maxillary hyperplasia, severe protrusion of the middle third of the face, and anterior displacement of the incisors, producing an appearance to the face historically referred as “Cooley’s face.”5
This study was performed to evaluate the trend of manifestations of the oro-maxillofacial changes in thalassemia major. The patients who took part in the research lived in southwest Iran.
METHODS AND MATERIALS
This descriptive cross-sectional study was performed on 119 blood-recipient thalassemic patients who predominately received inappropriate transfusion therapy. The patients were divided into six age groups including: ages 1 to 4; ages 5 to 6; ages 7 to 9; ages 10 to 12; ages 13 to 15; and over the age of 16. They were examined clinically for maxillofacial manifestations by two dental specialists separately, and photographs were analyzed respectively. If there was any difference in clinical observation, that patient was visited by a third specialist. The collected data were statistically analyzed by SPSS software (SPSS, Inc, Chicago, IL).
RESULTS
Of 119 examined thalassemic patients from ages 1 to 28 years old, 49.6% were male and 50.4% were female. The main findings of this study are described below.
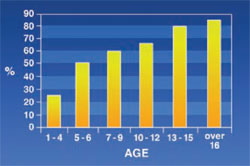
Maxillary Protrusion: Seen in 60.5% of the patients. This abnormality appeared to increase with age (Figure 1).
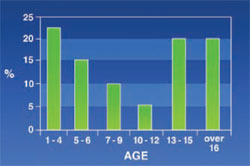
Mandibular Protrusion: Was not common in patients (5%). The highest incidence (8.3%) was found in the age group 7 to 9 (Figure 2).
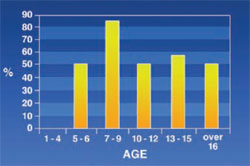
Saddle Nose: Was observed in 34% of the subjects. A positive correlation was found between this sign and age (Figure 3).
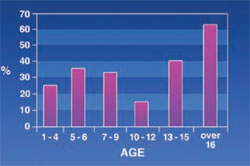
Spacing: 13.4% of the patients had spacing (more than two spaces is consider as spacing) (Figure 4). The incidence of this disorder decreased until the age of 12, then began to increase.
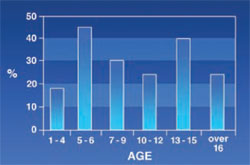
Posterior Malocclusion: Was seen in 27.7% of the patients. These abnormalities included cross bite, open bite, improper relation of molars, and other types of malocclusions. The posterior malocclusion had no certain pattern and its rate varied from one age group to another (Figure 5).
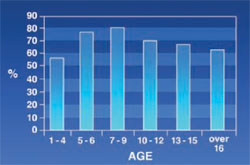
Anterior Malocclusion: 63% of the patients had anterior malocclusion, including open bite, cross bite, and edge-to-edge. Figure 6 shows that the rate of anterior malocclusion increased in the first 9 years of life and decreased afterward.
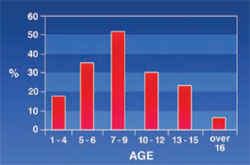
Lip Incompetency: 47.9% of the patients appeared to have lip incompetency increasing with age (Figure 7).
Oral Mucous Membrane: In 26.9% of the patients, the oral mucous membrane was pale. As can be seen in Figure 8, this discoloration of the mucous membrane increased until patients reached 7 to 9 years of age and decreased afterward.
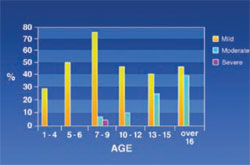
Gingival Discoloration: Severe gingival discoloration was seen in only 0.8% of the patients while mild and moderate discoloration was seen in 47% and 13% of the patients, respectively. It was shown that intensity and rate of gingival discoloration increased with age (Figure 9).
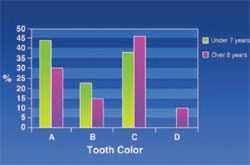
Tooth Color: Using a shade guide (Ivoclar Vivadent, Amherst, NY), the upper incisor was considered for tooth color in patients. Figure 10 shows that “D” color was absent in patients under 7 years old and “C” color was significantly lower in this age group compared with patients aged over 8 years.
Xerostomia and Oral Lesions: Xerostomia and oral lesions were not found in any of the patients during the time of the study.
DISCUSSION
The results of this study showed that the rate of maxillary protrusion increased with age in thalassemic patients. This was quite predictable because the maxilla is one of the most common bones affected by thalassemia.2 Results of the study were similar to results reported by Capurso, De Mattia, and Ba¸ssimitçi.6-8 It is generally accepted that osteal signs in thalassemic patients appear only after other signs materialize. However, in this study maxillary protrusion was seen even in the patients aged 1 to 4; this might have been a result of a non-intensive blood-transfusion program.
Reports from other studies show that mandibular protrusion is less prevalent than maxillary protrusion in thalassemic patients and this is mainly because of the firm cortex of the mandible.5 In this study the highest rate of mandibular protrusion (8%) occurred in 7- to 9-year-old patients while maxillary protrusion affected 80% of the patients.
Findings showed that the rate of saddle nose increased with age; this was predictable because the activity of bone marrow increases with age, giving a saddle shape to the nose.
The rate of spacing was found to be 12.5% altogether. Considering the signs of maxillofacial changes in thalassemic patients and bone expansion, spacing is likely to increase as thalassemic patients age.
Contrary to the authors’ expectations, it was found that spacing had an inverse correlation with age until age 12 and a direct correlation with age afterwards. The decrease of spacing in patients under the age of 12 might have been because of missing deciduous teeth and the eruption of permanent teeth, as permanent teeth are larger than deciduous teeth. These results agree with those reported by Ficarra and Halstead, who showed that spacing increased with age in thalassemic patients.9,10 Aga Hosseini reported in thalassemic patients a 23% spacing, which is much more than its rate in unaffected people.11
All types of malocclusion (ie, Angle’s class, open bite, cross bite, etc) were evaluated in this study. The frequency of posterior malocclusion varied in different age groups. Osteal changes and early extraction of teeth because of caries and misplacing eruption of permanent teeth may account for this. De Mattia reported that major thalassemia produced various and serious malocclusions (Angle’s class, deep bite, and open bite).6 Abu Alhaija reported that in thalassemic patients, the average ABN angle, which defines the maxillary–mandibular relationship, was significantly larger and mandibular base length (Ar-Gn) was significantly less than that in the control group.12
Anterior malocclusion was more prevalent in thalassemic patients and had increased with age. The highest prevalence rate was found in the 7- to 9-year-old age group and its prevalence decreased with age afterwards. Criteria for considering anterior malocclusion were overjet and overbite ranging from higher than 3 mm or lower than 1 mm, and also the presence of open bite, cross bite, and edge-to-edge. It is worth noting that overjet and overbite were less common in patients older than age 9 years, despite its increasing rate in infancy and childhood.
In this study, the inclination of anterior teeth was inverted in higher-age patients. This might have been a result of the pressure of the lip on anterior teeth (because the patient tried to close his/her mouth and this made the lip seal on rest position). This in turn induced linguo-version of the incisal edge of anterior teeth. These changes might have caused the overjet of patients to be in or close to the normal range and that was why anterior malocclusion rate was reduced in older patients.
Maxillary protrusion was not common in the early stages of life; therefore, the lip did not put much pressure on the teeth. Sahafiaun reported a high prevalence (32%) of overjet in thalassemic patients.13 Cannell, Ba¸ssimitçi, and Capurso reported an increasing overjet in thalassemic patients.7,8,14 Maxillary protrusion and lip incompetency increased with age and this result was acceptable because with growth of the maxilla, lip incompetency increased. The authors’ results show a decrease in nose and lip incompetency in the 10- to 12-year-old patients. The authors could not determine the reason for this drop. Because of the high level of bilirubin in their blood, the mucosal membranes of thalassemic patients are pale and lemon-colored.5 This study showed that the rate of mucosal discoloration increases until the age of 9 and then it begins to decline to 5% for patients over 16 years old. Precipitation of iron in the mucous might have been a reason for these changes. De Mattia reported that in 60 patients ages 6 to 26, only three patients had a pale oral mucosa membrane.6
The results of this study showed that discoloration of the gingiva was age-dependent. Moderate and severe pigmentation could be seen in older patients while mild pigmentation affected younger patients. Previous studies have not taken this point into account.
The results of this study showed that the color of permanent teeth was much darker compared with deciduous teeth in thalassemic patients. Calcification of the deciduous central germ begins at 3 to 4 months of life in the prenatal period and will end 2 months after birth.15 Calcification of the permanent central germ starts at 3 to 4 months of age and will continue until 4 to 5 years of age. Clinical manifestations of thalassemia appear about 8 months after birth. With regard to the lysis of red blood cells, an increase of the serum’s iron and sedimentation of iron in teeth tissue during the calcification of permanent teeth and darkness of permanent teeth in thalassemic patients are expected. The level of iron in the tooth tissue of thalassemic patients is five times higher than in unaffected people.5 That would explain the increasing rate of “C” and “D” color in patients older than 8 years old. Considering the results of this study, further investigations are necessary to reveal the many unknown facts about oro-maxillofacial signs in thalassemic patients.
References
1. DeBall S, Gordy FM. Homozygous beta thalassemia in African-American pediatric patient. J Clin Pediatr Dent. 1997;21(4):315-319.2. Kliegman RM, Behromon RE. Nelson Essential of Pediatrics. 4th ed. Saunders Co; 2002:662-663.
3. Fauci AS, Braunwald E, Isselbacher KJ, et al. Harrison ‘s Principles of Internal Medicine. 14th ed. New York, NY: Mc Graw Hill; 1998:634-650.
4. Haqshenas M, Zamani J. Thalassemia [in Persian]. 1st ed. Shiraz: Shiraz University of Medical Sciences Publishing Center; 1997:29.
5. Greenberg MS, Glick M, Burcet LW. Oral Medicine Diagnosis and Treatment. 10th ed. Philadelphia, Pa: B.C. Decker; 2002:435-436.
6. De Mattia D, Pettini PL, Sabato V, et al. Oromaxillofacial change in thalassemia major. Minerva Pediatr. 1996;48:11-20.
7. Bassimitçi S, Yücel-Ero×glu E, Akalar M. Effects of thalassaemia major on components of the craniofacial complex. Br J Orthod. 1996;23(2):157-162.
8. Capurso U. Stomatognathic change in beta thalassemia. Congress of the European Academy of Pediatric dentistry. Athens, Greece; 1994.
9. Ficarra G, Hansen LS, Beckstead JH, et al. Thalassemia diagnosis through facial distortion. Int J Oral Maxillofac Surg. 1987;16(2):227-231.
10. Halstead CL. Oral manifestations of hemoglobinopathies. A case of homozygous hemoglobin C disease diagnosed as a result of dental radiographic changes. Oral Surg Oral Med Oral Pathol. 1970;30(5):615-623.
11. Aga Hosseini F, Shanbody M. Evaluation of craniofacial abnormality in thalassemic patients who referred to Ali-Asghar hospital in Tehran [in Farsi]. Journal of Dentistry of Tehran University of Medical Sciences. 2000;13: 16-24.
12. Abu Alhaija ES, Hattab FN, Al-Omari MA. Cephalometric measurements and facial deformities in subject with beta-thalassemia major. Eur J Orthod. 2002;24:9-19.
13. Sahafian A, Adeleh S, Aminian N. Evaluation of osteal changes of face and teeth in 50 major thalassemic patients [in Farsi]. Journal of Dentistry Mashhad University of Medical Sciences. 1993;23-29.
14. Cannell H. The development of oral and facial signs in thalassemia major. Br Dent J. 1981;164:50-51.
15. McDonald RE, Avery DR. Dentistry for the Child and Adolescent. 7th ed. St Louis, MI: The C.V. Mosby Co; 2000;181-182.
SIDEBAR 1
COMMENTARY
While this article helps us understand the dental and facial effects of a genetic disease, thalassemia, and what clinical changes to appearance and physical conditions occur, hopefully it will heighten the reader’s awareness of other dental conditions that are genetically based that can affect the appearance and function of teeth. Thalassemia occurs as a result of one single gene abnormality that affects the appearance of the maxillofacial region. Patients with Cooley’s anemia, which is also known as beta (b)-thalassemia or Mediterranean anemia, have a blood disease characterized by abnormality of the skull and long bone, which results in an atypical appearance and can include dentally maxillary protrusion, spacing of teeth, and anterior and posterior malocclusion to a greater degree than the normal population. The facial, dental, and soft tissue appearance differences and the percent of the population that have this condition that have these differences are described and listed in this article. In most cases the dental conditions that this group have can be treated if desired. The patient as an adult needs to be aware that their genetic make-up can be passed on to their progeny. As with any genetic disorder genetic counseling is important.
What other dental-oral conditions are based upon genetic abnormalities? Patients with ectodermal dysplasia exhibit numerous missing teeth and what teeth do develop will have compromised root and crown formation. As children the teeth never form and for esthetic and functional reasons over their lifetime prosthetic treatment will include removable prostheses and they will be good candidates for implants. In some cases genetic abnormalities are seen as conditions of the teeth. Amelogenesis imperfecta is another genetic syndrome that has specific tooth abnormalities. Fourteen different types have been described and all exhibit abnormalities that include primary or permanent teeth that are unusually small, discolored, pitted or grooved, and prone to rapid wear and breakage. Dentinogenesis imperfecta (hereditary opalescent dentin) is an autosomal dominant genetic disorder that can contribute to tooth discoloration (most often a blue-gray or yellow-brown color) and translucency. Three different types have been described. Teeth are also weaker than normal, making them prone to rapid wear, fracture, and loss. Another genetically passed on trait is missing teeth. The most commonly seen example is congenitally missing maxillary lateral incisors.
When patients are seen with known genetically transferred dental anomalies, it is important that the clinician discuss this with the patient and/or parent and refer them to appropriate genetic counseling to better understand the future implications.
Ashraf Sayyedi, DDS, MS
Assistant Professor of Prosthodontics
Faculty of Medicine
Yasuj University of Medical Sciences
Yasuj, Iran
Fereydoun Pourdanesh, DDS, MS
Assistant Professor of Oral and Maxillofacial Surgery
Shahid Beheshti University of Medical Sciences
Tehran, Iran
Bahador Sarkari, PhD
Assistant Professor of Prosthodontics
Faculty of Medicine
Yasuj University of Medical Sciences
Yasuj, Iran
Sayyed Hesamedin Nabavizadeh, MD, MS
Assistant Professor of Pediatrics
Faculty of Medicine
Yasuj University of Medical Sciences
Yasuj, Iran
 |  | |
| Figure 1 Frequency of maxillary protrusion in thalassemic patients. | Figure 2 Frequency of mandibular protrusion in thalassemic patients. | |
 |  | |
| Figure 3 Frequency of saddle nose in thalassemic patients. | Figure 4 Frequency of spacing in thalassemic patients. | |
 |  | |
| Figure 5 Frequency of posterior malocclusion in thalassemic patients. | Figure 6 Frequency of anterior malocclusion in thalassemic patients. | |
 |  | |
| Figure 7 Frequency of lip incompetency in thalassemic patients. | Figure 8 Frequency of mucosal discoloration in thalassemic patients. | |
 |  | |
| Figure 9 Frequency of gingival pigmentation in thalassemic patients. | Figure 10 Frequency of tooth colors in thalassemic patients. |

