
What Will the Proposed Changes to the ADA’s Sedation Guidelines Mean for Patients and Practitioners?
Raymond A. Dionne, DDS, PhD | November 5, 2015
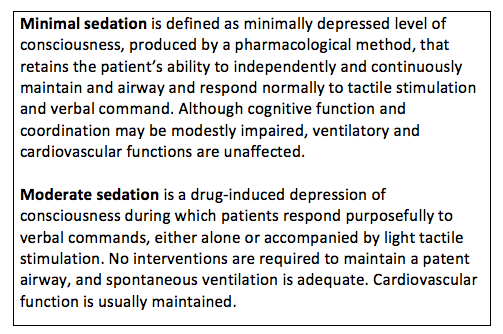
The proposed revisions of the ADA Guidelines for the Use of Sedation and General Anesthesia by Dentists differentiates levels of sedation (minimal or moderate; see sidebar) based on the administration of the maximum recommended dose (MRD) of the sedative-hypnotic drug being administered. Based on this classification scheme, the number of training hours, monitoring equipment, and the type of clinical training differ markedly and require varying levels of training in life support. The difference between training in minimal sedation (N=16 hours) and moderate sedation training (N=60 hours) is substantial and very likely will limit the number of dentists who can afford to be trained. Decreasing the dental manpower to provide sedation services will perpetuate fear of dentistry as a barrier to access to care for the substantial portion patients (approximately 15%) who report themselves as “terrified” or “very nervous” about receiving dental care (Dionne et al., JADA 1998). An underlying assumption in using the MRD to predict safety and the requisite levels of training is that the recommended dose is for sedation and based on scientific evidence and regulatory review by the Food and Drug Administration (FDA). However, most drugs that are used for sedation of dental outpatients are used off-label and have not been evaluated by the FDA for safety and effectiveness for this indication.
The Off-Label Use of Sedative-Hypnotics
The use of an approved drug in another dosage form, for another indication, at higher doses, in a different patient population, or for a use that is not in the original labeling is considered to be an off-label use. The FDA recognizes that the off-label use of drugs by practitioners often is appropriate and can result in therapeutic innovation and become a standard practice in the absence of regulatory approval for that use. For example, tricyclic antidepressants were recognized in the 1970s to be effective for relieving chronic pain at doses lower that those used to treat depression and eventually became widely used for the treatment of pain. Some of the drugs used for oral sedation in dentistry, such as chloral hydrate, were introduced as sedatives before it was required to show evidence of efficacy in well-controlled trials and demonstrate safety in extensive animal testing and carefully controlled dose escalation studies in humans. Introducing drugs that satisfied these requirements into clinical practice (eg, benzodiazepines) has resulted in drugs that are more selective for anxiety relief versus nonspecific CNS depression manifesting as sedation, respiratory depression, and decreased consciousness. Few drugs currently available in the United States have an approved indication for procedural sedation, making the use of most drugs and doses used by dentists for sedation to be non-validated, off-label use from a regulatory perspective.
Recommended Doses
The FDA is a regulatory agency within the federal government that is: “responsible for protecting the public health by assuring the safety, efficacy and security of human and veterinary drugs, biologic products medical devices, and the nation’s food supply, cosmetics and products that emit radiation.” As part of this process, the FDA approves the content of the package insert (also described as the “label”) as the official prescribing information. This label describes the approved indications for use of the drug and clinical pharmacology information including dosage, adverse reactions, and special warnings and precautions. A licensed clinician is not bound by the package insert and may legally prescribe a drug for any purpose that they deem appropriate. A clinician, however, may be vulnerable to litigation if adverse effects result from an unapproved use of a drug.
Demonstrating a drug’s efficacy to the FDA requires performing well-controlled clinical investigations. The determination of safety is more complex as no drug is totally safe: all drugs produce unwanted effects in some patients at therapeutic doses. Many serious adverse effects of drugs are infrequent and may go undetected in the relatively small samples used in clinical trials for drug approval, usually no more than a few thousand participants. The voluntary reporting systems in the United States are not as robust as legally mandated systems in some other countries (Rivera SM, Gilman AG. Drug intervention and the pharmaceutical industry. In: Brunton LL, Chabner BA, Knollman BC, eds. Goodman and Gilman’s The Pharmacological Basis of Therapeutics) and few clinicians file adverse drug reports. Moreover, estimates of the incidence of adverse events to predict safety cannot be quantified in the absence of any reliable estimates of adverse events and how many drug administrations occurred or what doses were used. As a consequence, determining what drugs and doses for an off-label indication are safe and effective is left to clinicians administering the drugs and only become matters of public concern when serious morbidity and mortality are reported in the news media and reflect poorly on the profession.
Determining the Relationship Between Dose and Patient Reponses
Individuals vary in the magnitude of their responses to the same dose of a drug or to a similar drug from the same class, and even the same individual may not respond similarly to the same drug dose at a subsequent administration. Variability in the pharmacodynamics response of the population is influenced by many factors including genetics, age, disease, body weight and the presence of co-administered drugs. This variability forms the basis for dose-response curves that illustrate the range of responses from a fixed dose that usually ranges over several orders of magnitude. The response to drugs administered for subjective symptoms such as dental pain and apprehension is highly variable and can range from not achieving a therapeutic effect at a fixed dose to developing significant morbidity or even mortality. For example, the range of morphine needed to achieve pain relief following general surgery varies 40-fold when patient-controlled analgesia is used.
The dose of a drug required to produce a specified effect (such as anxiety relief) in 50% of the population is calculated as the median effective dose. This dose does not predict the response of any given individual in the population but rather serves as a basis for comparing the relative efficacy of the drug to its median toxic dose. The ratio of these calculated median doses is a statement of the therapeutic window and is a reflection of a drug’s selectivity for producing its desired effect versus its adverse effects. For drugs used for sedation, this relationship defines the anxiolytic efficacy of the drug versus its risk of producing respiratory depression, the most common adverse effect that results in serious morbidity or mortality. Therapeutic success and safety result from integrating efficacy and safety from animal and clinical studies with individual factors that determine the individual response of a given patient. Extrapolating from a dose determined from population data, such as the maximum recommended dose, does not directly predict the safety of a drug in an individual patient, especially if based on data for another indication. A recent example of the hazards of extrapolating across indications and patient populations was the apparent safety of selective COX-2 inhibitors for short-term use for acute pain to their chronic administration to prevent the development of colon polyps. The unexpected findings of increased cardiovascular events and stroke when administered chronically resulted in their removal from the market and a ‘black box’ warning for virtually all COX2 inhibitors and NSAIDs.
Evidence-Based or Eminence-Based Guidelines to Improve the Safety of Sedation?
The dental profession recognized the need to standardize training in anesthesia and sedation for dentists with consensus development meetings, leading to the formulation of the Guidelines in the 1970s and subsequent revisions in reaction to continued public concerns. Three decades ago, the NIH Consensus Development Conference on Anesthesia and Sedation in the Dental Office called for more research to validate the safety of these procedures (Dionne RA and Laskin DM, Elsevier, 1986). A workshop on the safety of enteral sedation asserted the safety of this modality while also recommending more research to demonstrate the safety of individual drugs, doses, monitoring methods and training sufficient to assure competence (Dionne at al., JADA 2006). While at least one large-scale clinical trial has evaluated the safety of parenteral administration of sedative drugs and combinations (Dionne et al., JADA 2001), only case reports, case series (Gordon et al., Gen Dent. 2007) and a retrospective closed claims study from one dental specialty (Bennett et al., JADA 2015) provide very limited evidence for the safety of sedation and anesthesia by the dental profession.
Given the widespread recognition for basing dental practices on scientific evidence, why are we now considering a return to eminence-based clinical practices? The arbitrary use of the MRD for drugs approved for a different therapeutic indication, the induction of sleep, is a fallacious surrogate endpoint for predicting the safety of drugs used off-label for sedation, determining monitoring requirements and promulgating training requirements. The dental profession and their patients deserve policies for training and safety that are based on valid endpoints and an unbiased review of the literature by the ADA’s Center for Evidence-Based Dentistry. The dearth of scientific evidence upon which to support the current dental use of anxiolytic, adjunctive agents and anesthetics would be greatly enhanced by developing national statistics for morbidity and mortality that differentiates safe drugs and practices from problematic ones for all levels of anesthesia and sedation. If implemented, the proposed revisions of the Guidelines will limit the ability of general dentists to provide sedation services to patients who would otherwise avoid restorative and preventive dental procedures. This will herald the return to limited treatment options for fearful patients: extractions provided by a small group of specialists who have been trained in parenteral sedation.
About the Author
Raymond A. Dionne, DDS, PhD, is research professor in the department of pharmacology & toxicology at Brody School of Medicine and in the department of foundational sciences in the School of Dental Medicine at East Carolina University in Greenville, North Carolina. He can be reached at dionner@ecu.edu.

